FDA AI Medical Devices 2026: Startup Compliance Action Plan

Navigating the FDA’s New 2026 AI-in-Medical-Devices Guidance: A Startup’s 3-Month Action Plan for Compliance
The landscape of medical technology is evolving at an unprecedented pace, largely driven by the transformative power of Artificial Intelligence (AI). For startups at the forefront of this innovation, the promise of AI in medical devices is immense – from enhancing diagnostic accuracy to personalizing treatment plans and improving patient outcomes. However, this rapid advancement also brings with it a complex web of regulatory challenges, particularly from the U.S. Food and Drug Administration (FDA).
The FDA has been actively working to establish a robust regulatory framework for AI and Machine Learning (ML)-enabled medical devices (AI/ML-MDs). With new guidance anticipated for 2026, the clock is ticking for startups to align their development and deployment strategies with these impending regulations. Ignoring these changes is not an option; proactive compliance is the cornerstone of market access and sustained success. This article provides a comprehensive, practical 3-month action plan specifically designed for startups to navigate the FDA’s new 2026 AI-in-medical-devices guidance, ensuring they are not only compliant but also positioned for future growth.
Understanding the Evolving Landscape of FDA AI Medical Devices Regulation
Before diving into the action plan, it’s crucial to grasp the FDA’s current stance and the direction of future regulations concerning FDA AI medical devices. The FDA recognizes the unique characteristics of AI/ML-MDs, particularly their adaptive capabilities, which allow them to learn and change over time. This adaptability, while beneficial, presents significant challenges for traditional regulatory paradigms that typically evaluate fixed devices.
Historically, the FDA has released several foundational documents, including the discussion paper on ‘Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD)’ in 2019, followed by the ‘AI/ML-Based SaMD Action Plan’ in 2021. More recently, the FDA has emphasized the importance of ‘Good Machine Learning Practice (GMLP)’ principles and transparent reporting for AI/ML-MDs. The anticipated 2026 guidance is expected to consolidate these principles, offering clearer pathways for premarket submission, postmarket surveillance, and managing modifications to AI algorithms.
Key areas of focus for the FDA include:
- Predetermined Change Control Plans (PCCPs): For adaptive AI/ML-MDs, the FDA is moving towards requiring manufacturers to define and validate specific modifications that the algorithm can make within a predefined scope, without needing a new 510(k) or PMA submission for every minor change. This is a critical shift for dynamic AI systems.
- Transparency and Explainability: The FDA expects manufacturers to provide clear documentation on how their AI models work, their limitations, and the data used for training and validation. This includes understanding potential biases and ensuring robust performance across diverse patient populations.
- Real-World Performance Monitoring: Postmarket surveillance will be increasingly important, with an emphasis on monitoring the real-world performance of AI/ML-MDs to ensure their continued safety and effectiveness as they learn and adapt.
- Data Management and Cybersecurity: Given the data-intensive nature of AI, robust data governance, privacy, and cybersecurity measures are paramount to protect patient data and ensure device integrity.
For startups, these evolving regulations mean that a ‘set it and forget it’ approach to AI development is no longer viable. Compliance must be integrated into every stage of the product lifecycle, from initial design to postmarket deployment. The upcoming 2026 guidance for FDA AI medical devices will likely formalize many of these expectations, making proactive preparation essential.
The 3-Month Action Plan for FDA AI Medical Devices Compliance
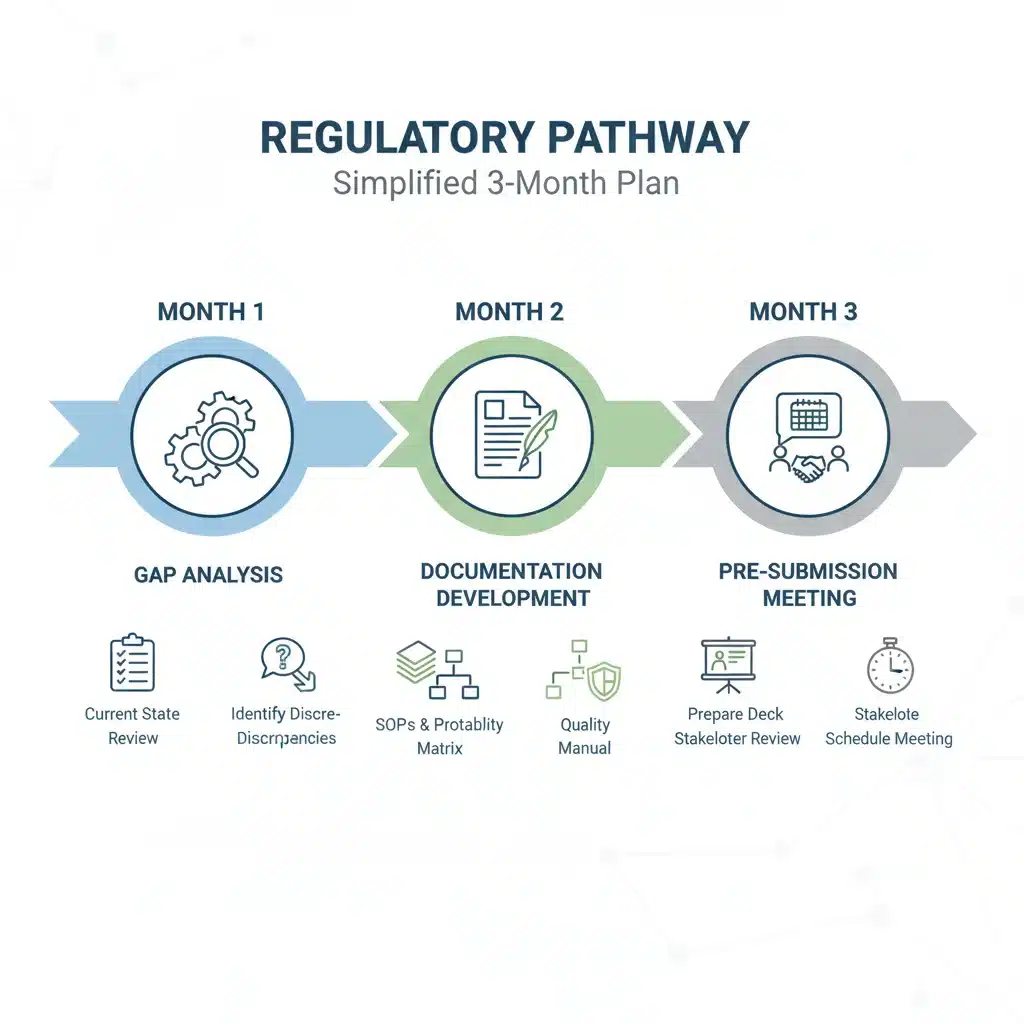
This action plan is structured to provide a clear, actionable roadmap for startups over a 90-day period. Each month focuses on distinct, yet interconnected, objectives to build a robust compliance framework for FDA AI medical devices.
Month 1: Assessment and Foundation Building
The first month is dedicated to understanding your current position relative to the anticipated FDA AI medical devices guidance and establishing the foundational elements for compliance.
Week 1-2: Regulatory Intelligence and Gap Analysis
- Deep Dive into FDA Guidance: Assign a dedicated team member (or consultant) to thoroughly review all available FDA guidance documents related to AI/ML-MDs, including the 2019 discussion paper, the 2021 action plan, and any recent workshops or presentations from FDA officials. Pay close attention to recent public comments or summaries of anticipated 2026 changes.
- Identify Applicable Regulations: Determine which specific regulatory pathways (e.g., 510(k), De Novo, PMA) are most likely to apply to your AI medical device. Understand the classification of your device (e.g., SaMD, medical device accessory).
- Internal Audit and Gap Analysis: Conduct a comprehensive audit of your current AI development processes, quality management system (QMS), and documentation practices against the identified FDA expectations. This includes:
- AI Model Development Lifecycle: How are your models designed, trained, tested, and validated? What data sources are used? Are there robust version control mechanisms?
- Data Governance: What are your data collection, storage, security, and privacy protocols (e.g., HIPAA compliance)? How do you ensure data quality and representativeness?
- Risk Management: Is there a formal risk management process in place that specifically addresses AI-related risks (e.g., algorithmic bias, model drift, cybersecurity vulnerabilities)?
- Postmarket Surveillance Plan: Do you have a strategy for monitoring device performance in the real world and managing potential modifications?
- Quality Management System (QMS): Assess whether your existing QMS (e.g., ISO 13485) adequately covers AI/ML-specific requirements.
- Documentation Review: Scrutinize existing documentation for completeness, clarity, and alignment with FDA principles. This includes design specifications, validation reports, risk assessments, and software development lifecycle (SDLC) documentation.
Week 3-4: Team Alignment and Resource Allocation
- Form a Dedicated Compliance Task Force: Assemble a cross-functional team comprising representatives from product development, data science, regulatory affairs, quality assurance, and legal. This team will be responsible for driving compliance efforts.
- Define Roles and Responsibilities: Clearly delineate who is responsible for each aspect of compliance, from documentation to technical implementation.
- Budget and Resource Allocation: Identify any external resources needed, such as regulatory consultants specializing in AI/ML-MDs, legal counsel, or specialized testing services. Allocate budget accordingly.
- Training and Education: Provide targeted training to relevant teams on FDA AI medical devices regulations, GMLP principles, and internal compliance procedures. Ensure everyone understands the implications of the 2026 guidance.
- Initial QMS Updates: Begin the process of updating your QMS to specifically incorporate AI/ML device development and management processes. This might include new procedures for data management, model validation, and change control.

Month 2: Strategy Development and Documentation Enhancement
Building on the foundation laid in Month 1, the second month focuses on refining your regulatory strategy and developing the necessary documentation to demonstrate compliance with FDA AI medical devices guidance.
Week 5-6: Predetermined Change Control Plan (PCCP) Development
- Develop Your PCCP Strategy: For adaptive AI/ML-MDs, this is paramount. Define the types of modifications your AI model might undergo (e.g., algorithm updates, retraining with new data) and establish a clear framework for how these changes will be managed.
- Define ‘Scope of Acceptable Change’: Clearly articulate the boundaries within which your AI model can adapt without requiring a new premarket submission. This includes defining performance metrics, safety limits, and data types allowable for retraining.
- Validation Protocol for Changes: Outline the specific testing and validation protocols that will be applied to any modifications made under the PCCP to ensure continued safety and effectiveness.
- Documentation of PCCP: Formalize the PCCP in a comprehensive document that details the rationale, methods, and validation strategy. This will be a key component of your FDA submission.
Week 7-8: Robust Data Management and AI Model Documentation
- Data Governance Framework: Implement or refine a robust data governance framework that covers data acquisition, curation, labeling, storage, security, and privacy. Ensure traceability of all data used for training and validation.
- Bias Mitigation Strategy: Develop and document a strategy for identifying, mitigating, and monitoring potential biases in your training data and AI model outputs. This includes diversity in datasets and fairness metrics.
- AI Model Transparency and Explainability: Prepare detailed documentation on your AI model’s architecture, training methodology, performance metrics, and limitations. Focus on making the model’s decision-making process as transparent and explainable as possible, commensurate with its risk.
- Validation and Verification (V&V) Plan: Create a comprehensive V&V plan that includes both retrospective and prospective studies, addressing performance across diverse patient populations and clinical scenarios. This plan should align with GMLP principles.
- Risk Management File Updates: Integrate AI-specific risks (e.g., algorithmic bias, model drift, cybersecurity threats to AI algorithms) into your risk management file (per ISO 14971), including mitigation strategies and residual risk assessment.
Month 3: Pre-Submission, Testing, and Final Preparations
The final month is about solidifying your submission readiness, performing critical tests, and engaging with the FDA.
Week 9-10: Independent Verification and Pre-Submission Meeting Preparation
- Independent Verification and Validation: Engage third-party experts or internal teams distinct from the development team to independently verify and validate your AI model’s performance, PCCP, and overall compliance. This adds credibility to your data.
- Simulated Clinical Performance Testing: Conduct rigorous simulated testing to evaluate your AI medical device’s performance under various clinical conditions, including edge cases and potential failure modes. Document all results meticulously.
- Cybersecurity Assessment: Perform a thorough cybersecurity assessment of your device, including penetration testing and vulnerability analysis, especially concerning AI model integrity and data protection.
- Prepare Pre-Submission Meeting Request: Draft a detailed pre-submission meeting request to the FDA. This document should clearly outline your device, its intended use, the regulatory pathway you anticipate, and specific questions you have for the FDA regarding your compliance strategy, particularly your PCCP and V&V plan for FDA AI medical devices.
- Internal Review of All Documentation: Conduct a final, comprehensive internal review of all regulatory documentation, including your QMS, technical documentation, risk management file, and proposed submission package. Ensure consistency, accuracy, and completeness.
Week 11-12: FDA Engagement and Final Readiness
- Submit Pre-Submission Meeting Request: Formally submit your pre-submission meeting request to the FDA. This is an invaluable opportunity to get direct feedback from the agency before a full submission, potentially saving significant time and resources.
- Develop Postmarket Surveillance Plan: Finalize your plan for continuous monitoring of your AI medical device’s performance in the real world. This includes strategies for detecting model drift, managing updates outside the PCCP, and collecting user feedback.
- Training for Postmarket Activities: Train relevant personnel on the postmarket surveillance plan, including adverse event reporting and managing device modifications.
- Refine Business Strategy: Based on the insights gained and the regulatory framework established, refine your business strategy for market entry and growth, considering the long-term implications of FDA AI medical devices compliance.
- Contingency Planning: Develop contingency plans for potential regulatory hurdles or unexpected findings during testing or FDA interactions.
Practical Solutions and Best Practices for Startups
Beyond the structured action plan, several practical solutions and best practices can significantly aid startups in navigating the complexities of FDA AI medical devices regulations:
- Embrace a Quality-by-Design Approach: Integrate regulatory requirements and quality considerations into the very earliest stages of AI product development. This prevents costly rework later on.
- Leverage Existing Standards: Utilize relevant industry standards and guidelines, such as ISO 13485 for quality management, ISO 14971 for risk management, and cybersecurity standards, adapting them for AI/ML specifics.
- Invest in Data Infrastructure: A robust, secure, and well-managed data infrastructure is critical. This includes data versioning, audit trails, and strong access controls.
- Prioritize Explainable AI (XAI): While not always fully achievable, striving for greater explainability in your AI models can significantly ease regulatory review and build trust.
- Modular Design for AI: Design your AI components with modularity in mind. This can make it easier to validate specific modules and manage changes under a PCCP.
- Engage with the FDA Early and Often: The pre-submission process is invaluable. Don’t hesitate to seek guidance from the FDA on novel aspects of your AI medical device.
- Stay Updated: The regulatory landscape for FDA AI medical devices is dynamic. Continuously monitor FDA announcements, workshops, and new guidance documents. Subscribe to FDA newsletters and industry publications.
- Build a Strong Internal Regulatory Culture: Foster an environment where regulatory compliance is seen as a shared responsibility, not just the domain of a single department.
- Consider Cloud Compliance: If deploying AI in the cloud, ensure your cloud provider meets medical device regulatory requirements for data storage, security, and computational integrity.
- Pilot Programs and Real-World Evidence: Explore opportunities for pilot programs or real-world evidence generation to demonstrate the safety and effectiveness of your AI medical device in actual clinical settings, which can strengthen your regulatory submissions.
Common Pitfalls to Avoid
Startups often face unique challenges, and being aware of common pitfalls can save significant time and resources:
- Underestimating Regulatory Complexity: Assuming AI medical devices can be regulated like traditional software is a critical error. The adaptive nature of AI introduces new regulatory considerations.
- Ignoring Data Quality and Bias: Poor data quality, insufficient data, or biased datasets can undermine the safety and effectiveness of your AI, leading to regulatory rejection.
- Lack of Robust Documentation: Inadequate or inconsistent documentation of your AI’s development, validation, and risk management processes is a major red flag for the FDA.
- Failing to Plan for Postmarket Changes: Not having a clear strategy for managing AI model updates and continuous learning post-market will lead to compliance issues.
- Delaying Regulatory Engagement: Waiting until the last minute to engage with the FDA or to address compliance gaps can result in significant delays to market entry.
- Over-relying on Technology Alone: While cutting-edge AI is exciting, regulatory compliance requires a holistic approach that integrates technology with robust quality management and regulatory processes.
- Inadequate Cybersecurity Measures: AI models are prime targets for cyberattacks. Neglecting cybersecurity can compromise patient data and device integrity, leading to severe regulatory consequences.
- Disregarding Human Factors: How clinicians and patients interact with your AI medical device is crucial. Poor usability or unclear outputs can lead to errors, which the FDA closely scrutinizes.
The Future of FDA AI Medical Devices and Startup Opportunities
The 2026 FDA guidance on AI in medical devices is not just a hurdle; it’s an opportunity. By establishing clear regulatory pathways, the FDA aims to foster innovation while ensuring patient safety. Startups that proactively embrace these regulations will gain a significant competitive advantage. They will build trust with healthcare providers and patients, attract investors confident in their regulatory maturity, and ultimately bring life-changing AI medical devices to market faster and more responsibly.
The future will see an increased emphasis on real-world performance, continuous learning systems, and transparent reporting. Startups that can effectively demonstrate the safety, effectiveness, and fairness of their adaptive AI solutions through rigorous validation and robust postmarket surveillance will be the leaders in this new era of digital health. The key is to view regulatory compliance not as a burden, but as an integral part of product quality and a driver of innovation.
Conclusion
The FDA’s upcoming 2026 guidance for AI-in-medical-devices marks a pivotal moment for health tech startups. Proactive engagement and a structured approach to compliance are non-negotiable for success. This 3-month action plan provides a detailed roadmap, guiding startups through critical assessment, strategy development, and pre-submission preparations. By meticulously addressing regulatory intelligence, gap analysis, PCCP development, robust data management, and early FDA engagement, startups can transform potential regulatory challenges into strategic advantages.
Embracing a quality-by-design philosophy, leveraging existing standards, and fostering a strong internal regulatory culture will ensure that your AI innovations meet the stringent requirements of the FDA. The journey to bringing groundbreaking FDA AI medical devices to market is challenging but incredibly rewarding. By following this comprehensive plan, your startup can navigate the regulatory landscape with confidence, ensuring compliance, accelerating market access, and ultimately contributing to a healthier future powered by responsible AI.





